Algorytm Google'a w 30 minut rozpykał sekwencję aminokwasów, z którą nie radzili sobie naukowcy. To przełom
Sztuczna inteligencja Google'a rozwiązała właśnie jeden z trudniejszych problemów, z którym od lat nie mogli poradzić sobie naukowcy. Chodzi o przewidywanie trójwymiarowych struktur białkowych.

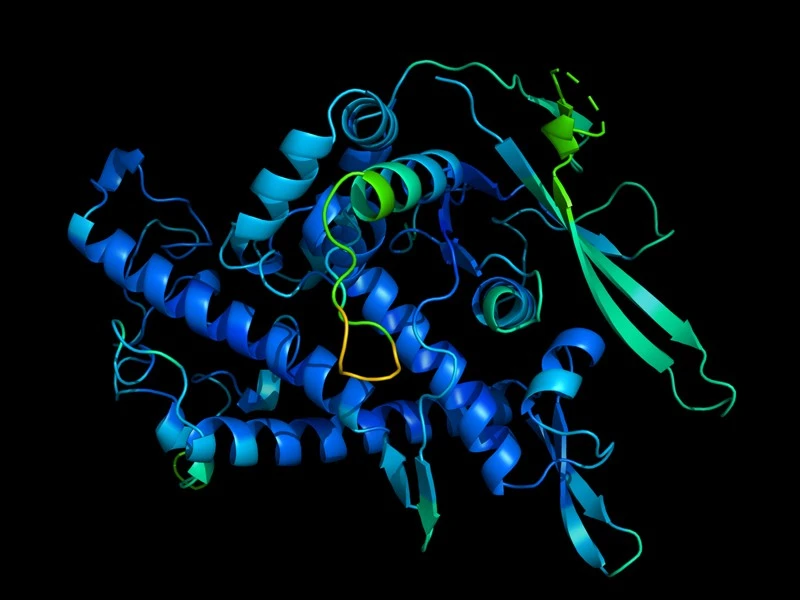
Opis problemu jest dość prosty: bierzemy daną sekwencję aminokwasów i na tej podstawie musimy rozrysować kształt jaki przyjmą białka z tej sekwencji. Możliwość określenia jak wyglądać będą takie struktury na podstawie ich sekwencji aminokwasów, przyspieszyłaby znacznie chociażby projektowanie nowych leków. Do niedawna jednak nie wiedzieliśmy jak to zrobić.
Od czego jednak są zaawansowane algorytmy
AlphaFold - algorytm opracowany przez naukowców z DeepMind, po przeanalizowaniu milionów sekwencji aminokwasów nauczył się bezbłędnego przewidywania struktur białkowych. Sztuczka ta pomogła AlphaFoldowi wygrać konkurs Critical Assessment of Structure Prediction, który odbywa się co dwa lata i którego celem jest wyłonienie najlepszego systemu zdolnego określać trójwymiarowe struktury białek.
Struktury przewidziane przez Alphafold, po porównaniu ich z wynikami prac laboratoryjnych okazały się bezbłędne. Jury konkursowe zaczęło podejrzewać wręcz zespół DeepMind o próbę jakiegoś zaawansowanego oszustwa.
Żeby to zweryfikować, Andrei Lupas, biolog z Instytutu Biologii Rozwojowej im. Maxa Plancka w Tybindze w Niemczech, który oceniał wyniki w CASP zlecił drużynie Alphafold dodatkowe zadanie. Polegało ono na stworzeniu struktury białka błonowego pewnej grupy drobnoustrojów, z którą zespół Lupasa nie mógł sobie poradzić od 10 lat. Algorytm DeepMind zwrócił dokładną budowę trzyczęściowego białka, stworzoną na podstawie sekwencji aminokwasów w… 30 minut.
To wielki przełom
Tak dokładne przewidywanie struktur białkowych oznacza, że algorytm DeepMind będzie w stanie znacząco przyspieszyć chociażby tworzenie nowych leków. Nie mówiąc już o tysiącach badań naukowych, które dzięki zaawansowanej analizie algorytmu prowadzone będą po prostu szybciej.
Problem z przewidywaniem struktur białkowych towarzyszy naukowcom od mniej więcej 50 lat. Ogromna liczba potencjalnych konfiguracji łączenia się białek sprawiała, że niektórzy przedstawiciele środowiska naukowego twierdzili, że zbadanie wszystkich molekularnych układów jest wręcz niemożliwe. Dlatego do tej pory ograniczaliśmy się do rozkodowywania tych struktur przy użyciu metod eksperymentalnych - krystalografii rentgenowskiej, czy też krioelektronowej mikroskopii.
Obie te metody są skuteczne, tyle że zajmują strasznie dużo czasu i nie zawsze udaje się uzyskać dzięki nim właściwy wynik. Niemniej jednak to właśnie dzięki metodom eksperymentalnym udało nam się opracować i poznać funkcje 170 tysięcy białek, z których zbudowane jest życie na naszej planecie. Niby dużo, jednak do przebadania nadal zostaje ponad 200 milionów.
A po co je badać? Zmapowanie wszystkich struktur białkowych pozwoli nam chociażby na poznanie ich funkcji. Nie jest to jednak najważniejsza rzecz. Ta wiedza przyda się zarówno przy projektowaniu nowych leków, których skuteczność w dużej mierze zależy od tego jak tworzące leki substancje są w stanie wpasować się w szczeliny i zakamarki danej struktury białkowej.
Niewykluczone też, że poznając coraz to więcej niuansów związanych z funkcjami przypisanymi do danego wyglądu struktury, będziemy w stanie tworzyć zupełnie nowe białka i wykorzystywać je w najróżniejszym celu. Od enzymów zdolnych do przyspieszonego rozkładu tworzyw sztucznych, aż po takie, które byłyby w stanie tworzyć dla nas biopaliwa przy minimalnym nakładzie energii. To oczywiście na razie tylko domysły, które w niedalekiej sprawdzi dla nas AlphaFold.